
High-throughput sequencing technology is a revolutionary change to traditional sequencing, sequencing hundreds of thousands to millions of DNA molecules at a time, so in some literature it is called next generation sequencing. The epoch-making changes, along with high-throughput sequencing, make it possible to perform detailed analysis of the transcriptome and genome of a species, so it is also known as deep sequencing.
The birth of high-throughput sequencing technology can be said to be a landmark event in the field of genomics research. This technology dramatically reduces the cost of single-base sequencing of nucleic acid sequencing compared to first-generation sequencing technologies. For example, human genome sequencing at the end of the last century cost $3 billion to decode human life codes, while second-generation sequencing The sequencing of the human genome has entered the era of the 10,000 genome. Such low cost of single base sequencing allows us to implement genome projects for more species to decrypt the genomic genetic code of more biological species. At the same time, large-scale whole-genome resequencing of other species of the species has become possible in species that have completed genome sequencing.
Summary and explanation of common nouns in the field of high-throughput sequencing
What is high-throughput sequencing?High-throughput sequencing (HTS) is a revolutionary change to traditional Sanger sequencing (called first-generation sequencing technology), sequencing hundreds of thousands to millions of nucleic acid molecules at a time, so in some literature It is called the next generation sequencing (NGS), which shows its epoch-making changes. At the same time, high-throughput sequencing makes it possible to analyze the transcriptome and genome of a species, so it is called deep sequencing. (Deep sequencing).
What is Sanger sequencing (first generation sequencing)
Sanger sequencing uses a DNA polymerase to extend primers that bind to a template of a sequence to be determined. Until a chain termination nucleotide is incorporated. Each sequence consists of a set of four separate reactions, each containing all four deoxynucleotide triphosphates (dNTPs) and spiked with a different amount of a different dideoxynucleoside triphosphate (ddNTP). Since the ddNTP lacks the 3-OH group required for extension, the extended oligonucleotide is selectively terminated at G, A, T or C. The termination point is determined by the corresponding dideoxy in the reaction. The relative concentrations of each of the dNTPs and ddNTPs can be adjusted to provide a set of chain termination products ranging from a few hundred to several kilobases in length. They have a common starting point, but terminate in different nucleotides. High-resolution denaturing gel electrophoresis can be used to separate fragments of different sizes. After gel treatment, X-ray film can be autoradiographed or non-isotopically labeled. Detection.
What is Genome Re-sequencing?
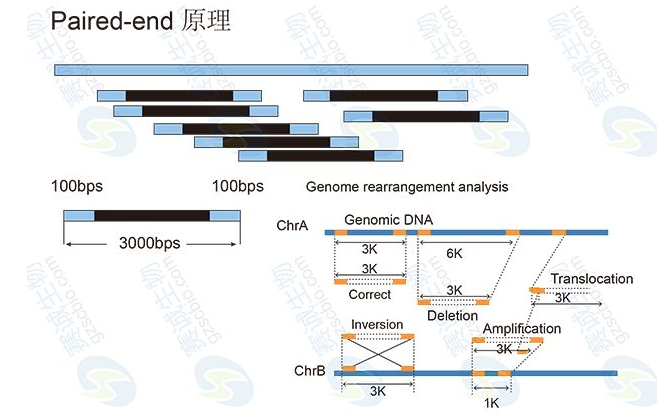
Whole genome resequencing is a method of performing genome sequencing on individuals with known genome sequences and performing differential analysis at the individual or population level. As the cost of genome sequencing continues to decrease, disease-causing mutation studies of human disease have expanded from exon regions to genome-wide. High-throughput sequencing by constructing libraries of inserts of different lengths combined with short-sequence, double-end sequencing, enabling detection of common, low-frequency, and even rare mutations in disease associations at the genome-wide level, as well as structural variations Etc., with significant scientific research and industrial value.
What is de novo sequencing
De novo sequencing is also known as de novo sequencing: it can sequence a species without any existing sequence data, and sequence and assemble the sequence using bioinformatics analysis to obtain the genomic map of the species. Obtaining a genome-wide sequence of a species is an important shortcut to speeding up understanding of this species. With the rapid development of next-generation sequencing technology, the cost and time required for genome sequencing are much lower than traditional technologies. Large-scale genome sequencing is getting better, and genomics research is also ushered in new development opportunities and revolutionary breakthroughs. Using a new generation of high-throughput, high-efficiency sequencing technologies and powerful bioinformatics capabilities, the genome sequences of all organisms can be measured and analyzed efficiently and cost-effectively.
What is external exon sequencing?
Exome sequencing refers to a genomic analysis method that uses sequence capture technology to capture and enrich the whole genome exon region DNA for high-throughput sequencing. Exon sequencing has lower cost than genomic resequencing, and has great advantages for studying SNPs and Indels of known genes, but it is impossible to study genomic structural variation such as chromosome break recombination.
What is mRNA sequencing (RNA-seq)
Transcriptomics is an emerging discipline after genomics that studies the type and copy number of all RNA (including mRNA and non-coding RNA) that a particular cell can transcribe under a certain functional state. Illumina's mRNA sequencing technology enables a variety of related research and new discoveries throughout the mRNA field. mRNA sequencing does not design primers or probes and is free to provide objective and authoritative information about transcription. Researchers can quickly generate complete poly-A tail RNA complete sequence information in a single experiment and analyze gene expression, cSNP, brand new transcription, brand new isoforms, splice sites, allele-specific expression and rare The most comprehensive transcriptome information such as transcription. Simple sample preparation and data analysis software supports mRNA sequencing studies in all species.
What is small RNA sequencing
Small RNA (micro RNAs, siRNAs and pi RNAs) are important regulators of life activities and play an important role in physiological processes such as regulation of gene expression, development of organisms, metabolism and disease. Illumina enables deep sequencing and quantitative analysis of all Small RNA in cells or tissues. In the experiment, the Small RNA in the range of 18-30 nt was first isolated from the total RNA, and the specific ends were added to the two ends, and then reverse-transcribed in vitro to prepare cDNA for further processing. Then the DNA fragment was subjected to unidirectional end using a sequencer. Direct sequencing. Through Illumina's large-scale sequencing analysis of Small RNA, the genome-wide miRNA map of the species can be obtained, including the excavation of new miRNA molecules, the prediction and identification of target genes, differential expression analysis between samples, miRNAs clustering and expression profiling. Analytical and other scientific applications.
What is miRNA sequencing?
Mature microRNA (miRNA) is a 17~24 nt single-stranded non-coding RNA molecule that interacts with mRNA to affect the stability and translation of target mRNA, ultimately induces gene silencing, and regulates biological processes such as gene expression, cell growth, and development. . Based on microRNA sequencing of second-generation sequencing technology, millions of microRNA sequences can be obtained at one time, which can quickly identify known and unknown microRNAs and their expression differences in different tissues, different developmental stages, and different disease states. It provides a powerful tool for the effects of cellular processes and their biological effects.
What is Chip-seq
Chromatin Immunoprecipitation (ChIP), also known as binding site analysis, is a powerful tool for studying the interaction between proteins and DNA in vivo. It is commonly used in the study of transcription factor binding sites or histone-specific modification sites. The ChIP-Seq technology, which combines ChIP with second-generation sequencing technology, efficiently detects DNA segments interacting with histones, transcription factors, etc., across the genome.
The principle of ChIP-Seq is: firstly, the DNA fragment of the target protein is specifically enriched by chromatin immunoprecipitation (ChIP), purified and constructed by the library; then the ligated DNA fragment is Qualcomm. Sequencing. By precisely mapping the millions of sequence tags obtained to the genome, the researchers obtained DNA segment information that interacts with histones, transcription factors, etc. across the genome.
What is CHIRP-Seq
CHIRP-Seq (Chromatin Isolation by RNA Purification) is a high-throughput sequencing method for detecting RNA-bound DNA and proteins. The method is to design a biotin or streptavidin probe to pull down the target RNA, and then the DNA fragment that interacts with it will be attached to the magnetic beads, and finally the chromosome fragments will be subjected to high-throughput sequencing. It is obtained which regions of the genome the RNA can bind to, but because the protein sequencing technology is not mature enough, it is impossible to know the protein bound to the RNA.
What is RIP-seq
RNA Immunoprecipitation is a technique for studying the binding of RNA to proteins in cells. It is a powerful tool for understanding the dynamic processes of post-transcriptional regulation networks and can help us discover regulatory targets for miRNAs. This technique uses an antibody against a target protein to precipitate the corresponding RNA-protein complex, and then isolates and purifies the RNA bound to the complex.
RIP can be seen as a similar application of the commonly used chromatin immunoprecipitation ChIP technique, but since the study object is an RNA-protein complex rather than a DNA-protein complex, the optimization conditions of the RIP experiment are not the same as those of the ChIP experiment (such as compounding). The substance does not need to be immobilized, and the reagents and antibodies in the RIP reaction system must not contain RNase, and the antibody needs to be verified by RIP test, etc.). RIP technology downstream combined with microarray technology called RIP-Chip helps us understand the overall level of RNA changes in cancer and other diseases.
What is CLIP-seq
CLIP-seq, also known as HITS-CLIP, is a cross-linking-immunprecipitation and high-throughput sequencing, which reveals the interaction of RNA molecules with RNA-binding proteins at the genome-wide level. Revolutionary technology. The main principle is based on the coupling of RNA molecules and RNA-binding proteins under ultraviolet irradiation, and the RNA-protein complexes are precipitated by specific antibodies of RNA-binding proteins, and the RNA fragments are recovered, and a linker, RT-PCR, etc. are added. In the step, high-throughput sequencing of these molecules is carried out, and then bioinformatics analysis, processing, summarization, and mining of specific laws, thereby revealing the regulation of RNA-binding proteins and RNA molecules and their significance to life.
What is metagenomic:
The Magenomics study targets the entire microbial community. Compared with traditional single bacterial research, it has many advantages, among which two important points are: (1) Microorganisms are usually symbiotic in a small habitat in a community, and many of their characteristics are based on the entire community environment and between individuals. Interacting, therefore, the Metatagenomics study is more likely to find its characteristics than a single individual study; (2) Metagenomics studies can isolate microorganisms that cannot be isolated and cultured in the laboratory without the need to isolate individual bacteria.
Metagenomics is an emerging scientific research direction in genomics. Metagenomics (also known as metagenomics, environmental genomics, ecogenomics, etc.) is the study of genomic genetic material extracted directly from environmental samples. Traditional microbial research relies on laboratory culture, and the rise of the metagenome fills a gap in microbiology research that cannot be cultured in traditional laboratories. Advances in DNA sequencing technology and improvements in sequencing throughput and analytical methods have led to a glimpse of this unknown genomic science field over the past few years.
What is SNP, SNV (single nucleotide site variation)
Single nucleotide polymorphism, single nucleotide polymorphism, SNP or single nucleotide position variation SNV. Polymorphisms caused by single nucleotide variations (substitutions, insertions or deletions) at the same position in the genomic DNA sequence between individuals. There is a difference in the single nucleotides at the same position in different species and individual genomic DNA sequences. Loci, DNA sequences, etc. with such differences can be used as markers for genome mapping. On average, approximately one nucleotide polymorphism may occur per 1000 nucleotides in the human genome, and some of the single nucleotide polymorphisms may be associated with disease, but most may be unrelated to the disease. Single nucleotide polymorphism is an important basis for studying the genetic variation of human families and animal and plant lines. When studying cancer genomic variation, a specific single nucleotide variation in cancer is a somatic mutation, called SNV, relative to normal tissue.
What is INDEL (genomic small fragment insertion)
An insertion or deletion of a small fragment ("50 bp") on the genome, similar to SNP/SNV.
What is copy number variation (CNV): genomic copy number variation
Genomic copy number variation is a form of genomic variation that typically results in an abnormal copy number of large fragments of DNA in the genome. For example, the normal chromosome copy number of human is 2, and the copy number of some chromosomal regions becomes 1 or 3. Thus, the copy number is deleted or increased in this region, and the gene expression amount in the region is also affected. If a chromosome is divided into four regions of ABCD, the amplification and deletion of the C region occur in ABCCD/ACBCD/ACCBCD/ABD, respectively. The position of amplification may be continuous amplification such as ABCCD or amplification at other positions. Such as ACBCD.
What is structure variation (SV): genomic structural variation
Chromosomal structural variation refers to the variation of large fragments that occur on a chromosome. It mainly includes the insertion and deletion of large fragments of chromosomes (causing changes in CNV), the flipping of a certain area inside the chromosome, and the inter-chromosome trans-location between the two chromosomes. The general SV display uses Circos software.
What is Segment duplication?
Generally referred to as the SD region, tandem repeats consist of a series of DNA fragments that are similar in sequence. Tandem repeat plays an important role in primate genes of human genetic diversity. On human chromosomes Y and 22, there are large SD sequences.
What is genotype and phenotype
Both genotype and phenotype; generally refers to the relationship between certain single nucleotide site variation and expression.
What is Read?
Sequence tags generated by high-throughput sequencing platforms are called reads.
What is soft-clipped reads
When a certain segment of the genome is deleted, or the transcriptome is spliced, during the sequencing process, when the reads across the deletion site and the splice site are replied to the genome, a reads are cut into two segments and matched to different regions. The reads are called soft-clipped reads, and these reads play an important role in identifying chromosome structural variations and foreign sequence integration.
What is multi-hits reads
Since most of the sequencing results in shorter reads, one reads can match multiple locations in the genome and cannot distinguish the location of its true source. Some tools are based on statistical models, such as assigning such reads to areas with more reads.
What is Contig?
The splicing software is based on the overlap area between the reads, and the spliced ​​sequence is called Contig (contig).
What is Scaffold?
Genomic de novo sequencing, after obtaining Contigs by reading splicing, it is often necessary to construct a 454 Paired-end library or an Illumina Mate-pair library to obtain sequences at both ends of a certain size fragment (such as 3Kb, 6Kb, 10Kb, 20Kb). Based on these sequences, it is possible to determine the order relationship between some Contigs, which are known as Contigs to form Scaffold.
What is the Contig N50?
Reads will get some Contigs of different lengths after splicing. Adding all Contig lengths together gives you a total Contig length. Then sort all Contigs from long to short, such as Contig 1, Contig 2, Contig 3. . .........Contig 25. Contig is sequentially added in this order. When the added length reaches half of the total length of Contig, the last Contig length added is Contig N50. Example: Contig 1+Contig 2+ Contig 3+Contig 4=Contig total length * 1/2, the length of Contig 4 is Contig N50. Contig N50 can be used as a criterion for the quality of genomic stitching.
What is the Scaffold N50?
The Scaffold N50 is similar to the definition of the Contig N50. Contigs splicing assembly to obtain some different lengths of Scaffolds. Adding all the Scaffold lengths together gives you a total Scaffold length. Then sort all the Scaffolds from long to short, such as Scaffold 1, Scaffold 2, Scaffold 3. . .........Scaffold 25. The Scaffold is sequentially added in this order. When the added length reaches half of the total length of the Scaffold, the length of the last added Scaffold is Scaffold N50. Example: Scaffold 1+Scaffold 2+ Scaffold 3 +Scaffold 4 +Scaffold 5=Scaffold total length * 1/2, the length of Scaffold 5 is Scaffold N50. Scaffold N50 can be used as a criterion for the quality of genomic stitching.
What is sequencing depth and coverage?
The depth of sequencing refers to the ratio of the total number of bases sequenced to the size of the genome to be tested. Assuming a gene size of 2M and a sequencing depth of 10X, the total amount of data obtained is 20M. Coverage refers to the proportion of sequences obtained by sequencing to the entire genome. Due to the existence of complex structures such as high GC and repetitive sequences in the genome, the sequences obtained by sequencing the final splicing assembly often cannot cover the region, and the region not obtained in this part is called Gap. For example, a bacterial genome is sequenced with a coverage of 98%, and 2% of the sequence regions are not obtained by sequencing.
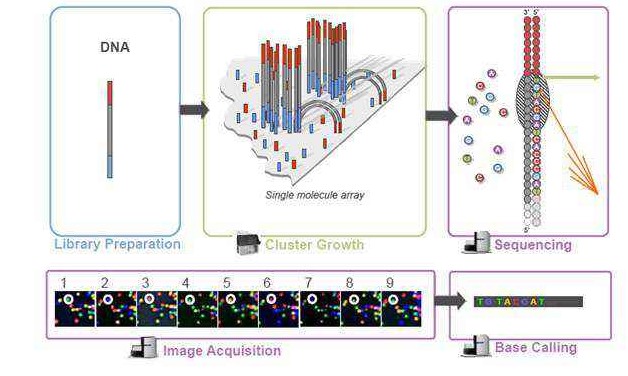
What is RPKM, FPKM
RPKM, Reads Per Kilobase of exon model per Million mapped reads, is defined in thisway [Mortazavi et al., 2008]:
The number of reads per 1K bases of the exon per 1 million maps of reads.
If there are 1 million reads mapped to the human genome, how many maps are there for each exon, and the length of the exons is different, so how many per 1K bases? The reads map is on, which is probably the intuitive explanation of this RPKM.
If it corresponds to a specific gene, then every 1 000000 of the reads mapped to the gene per kb is the read of the exon mapped to the gene.
Total exon reads:This is the number in the column with header Total exonreads in the row for the gene. This is the number of reads that have been mapped to a region in which an exon is annotated for the gene or across theboundaries of two exons or An intron and an exon for an annotated transcript of the gene. For eukaryotes, exons and their internal relationships are defined by annotations of type mRNA. Map the total number of reads on the exon. This is the number of reads mapped to a region that is either a known annotated gene or a boundary across two exons or an intron or exon of a transcript that has been annotated by a gene. For eukaryotes, the relationship between exons and their own internal is annotated by certain types of mRNA.
Exonlength: This is the number in the column with the header Exon length in the row for the gene, divided by 1000. This is calculated as the sum of the lengths of all exons annotated for the gene. Each exon is included only once inthis sum, even If it is present in more annotated transcripts for the gene.Partly overlapping exons will count with their full length, even though they share the same region. In the calculation, the sum of the lengths of all exons that have been annotated for all genes is calculated. Even if a gene is presented in multiple annotated transcripts, this exon is only included once in the summation. Even if partially overlapping exons share the same region, overlapping exons are calculated as their total length.
Mapped reads: The sum of all the numbers in the column with header Totalgene reads. The Total gene reads for a gene is the total number of reads that after mapping have been mapped to the region of the gene. Thus this includess all the reads uniquely mapped to The region of the gene as well asthose of the reads which match in more places (below the limit set in thedialog in figure18.110) that have been allocated to this gene's region. A gene's region is that comprised of the flanking regions(if it was Specified in figure 18.110), the exons, the introns and across exon-exon boundaries of all transcripts annotated for the gene. Thus, the sum of the total gene containing numbers is the number of mapped reads for the sample (you can find the number in the RNA-Seq report) The sum of the .map reads. Maps to the total number of all reads on a gene. So this contains all the only reads that are mapped to this area.
For example, if there are 1000 reades corresponding to the gene, the total number of reads is 1 million, and the total length of the exon of the gene is 5 kb, then its RPKM is: 10^9*1000 (reads)/10 ^6 (total number of reads) * 5000 (exon length) = 200 or: 1000 (reads) / 1 (million) * 5 (K) = 200 This value reflects the expression level of the gene.
FPKM (fragments per kilobase of exon per million fragments mapped). FPKM and RPKM calculation methods are basically the same. The difference is that FPKM calculates fragments, while RPKM calculates reads. Fragment has a broader meaning than read, so FPKM contains a broader meaning, either a pair-end fragment or a read.
What is transcript refactoring?
The sequenced data was assembled into a transcript. There are two ways to assemble: 1, de-novo construction; 2, with reference genome reconstruction. The de-novo assembly means that the overlapped reads are connected into a longer sequence without relying on the reference genome, and are continuously extended to form a contig and a scaffold. Common tools include velvet, trans-ABYSS, Trinity, etc. Reference genomic reconstruction refers to first posting the read back to the genome, and then obtaining the transcript in the genome through the read coverage, the information of the junction site, etc. Common tools include scripture and cufflinks.
What is genefusion?
Some or all of the two genes with different genomic positions are integrated to form a new gene, called a fusion gene, or a chimeric gene. It is possible for this gene to translate a fusion or chimeric protein.
What is the expression spectrum?
Gene expression profile: refers to the non-biased cDNA library of cells or tissues under a certain state, large-scale cDNA sequencing, collecting cDNA sequence fragments, qualitatively and quantitatively analyzing the mRNA population composition, thereby depicting the The gene expression profile and abundance information of a particular cell or tissue in a particular state, such that the data sheet is called a gene expression profile.
What is functional genomics?
Functional genomics (Functuionalgenomics), often referred to as Postgenomics, uses the information and products provided by the structural genome to develop and apply new experimental tools to comprehensively analyze gene function at the genomic or systemic level. It allows biological research to be systematically studied from the study of a single gene or protein to multiple genes or proteins. This is a biological functional study of genomic dynamics after the genomic static base sequence has been clarified. The research includes gene function discovery, gene expression analysis and mutation detection. Gene functions include: biological functions such as phosphorylation of specific proteins as protein kinases; cytological functions such as involvement in intercellular and intracellular signaling pathways; and developmental functions such as participation in morphogenesis. The methods used include classical subtractive hybridization, differential screening, cDNA representation of differential analysis, and mRNA differential display. However, these techniques cannot comprehensively and systematically analyze genes, and new techniques have emerged, including systematic analysis of gene expression (serial). Analysis of gene expression, SAGE), cDNA microarray, DNA chip and sequence tagged fragments display.
What is comparative genomics?
Comparative Genomics is a discipline that compares known genes and genomic structures based on genomic profiling and sequencing to understand gene function, expression mechanism, and species evolution. Using sequence and structural homology between the pattern organism genome and the human genome, clone human disease genes, reveal gene functions and molecular mechanisms of disease, elucidate the evolutionary relationship of species, and the internal structure of the genome.
What is epigenetics?
Epigenetics is a branch of genetics in which a gene expresses a heritable change without altering the nucleotide sequence of the gene. There are many epigenetic phenomena, known as DNA methylation, genomic impriting, maternal effects, genesilencing, nucleolar dominant, dormant transposon activation, and RNA editing. (RNA editing) and the like.
What is computational biology?
Computational biology refers to the development and application of data analysis and theoretical methods, mathematical modeling, computer simulation techniques, and so on. At present, the amount and complexity of biological data is increasing, and the data generated by genetic research every four months will double, and it is difficult to cope with observation and experiment alone. Therefore, we must rely on large-scale computational simulation technology to extract the most useful data from massive information.
What is genomic imprinting?
Genomic imprinting (also known as genetic imprinting) refers to the different expression of genes depending on the parent. The presence of an imprinted gene can result in one expression of two alleles in the cell while the other does not. Genomic imprinting is a normal process that has been found in some lower animals and plants for many years. Imprinted genes account for only a minority of the human genome, probably no more than 5%, but play a vital role in fetal growth and behavioral development. Genomic imprinting diseases mainly manifest as overgrowth, growth retardation, mental retardation, and behavioral abnormalities. At present, in the study of tumors, the deletion of imprint is considered to be one of the most common genetic factors causing tumors.
What is genomics?
Genomics (English genomics), a study of biological genomes and how to use genes. Used to generalize the branch of genetics involved in gene mapping, sequencing, and analysis of entire genome functions. The discipline provides genomic information and the use of relevant data systems to address major issues in the biological, medical, and industrial fields.
What is DNA methylation?
DNA methylation refers to the covalent bond of a methyl group at the 5' carbon position of the cytosine of the genomic CpG dinucleotide under the action of a DNA methyltransferase. Under normal circumstances, the CpG dinucleotide of the human genome "junk" sequence is relatively rare and always in a methylated state. In contrast, the human genome is about 100-1000 bp in size and is rich in CpG dinucleotides. The CpG island is always unmethylated and associated with 56% of the human genome coding genes. The results of human genome sequence analysis show that the human genome has about 28,890 CpG islands, and most chromosomes have 5-15 CpG islands per 1 Mb. The average value is 10.5 CpG islands per Mb, and the number of CpG islands is There is a good correspondence between gene density [9]. DNA methylation has become an important research in epigenetics and epigenomics due to the close relationship between DNA methylation and human development and tumor diseases, especially the transcriptional inactivation of tumor suppressor genes caused by methylation of CpG islands. content.
What is a genome annotation?
Genomeannotation is a hotspot of current functional genomics research using bioinformatics methods and tools to perform high-throughput annotation of the biological functions of all genes in the genome. The research of genome annotation includes two aspects: gene recognition and gene function annotation. The core of gene recognition is to determine the exact location of all genes in a genome-wide sequence.

The 60W Macbook Charger with MagSafe1 or Magsafe 2 Power Adapter has a magnetic DC connector, so if someone trips on it, the power cord disconnects harmlessly, keeping your MacBook Air safe. It also helps prevent the cable from fraying or weakening over time. Additionally, the magnetic DC helps guide the plug into the system for a quick and safe connection.
60W Apple charger usb c,60w charger macbook air,macbook 60w charger
Shenzhen Waweis Technology Co., Ltd. , https://www.szwaweischarger.com